Síndrome de Apert

¿Qué es el SÍNDROME DE APERT?
De nombre científico Acrocefalosindactilia tipo 1, el síndrome de Apert se engloba en un grupo de enfermedades como son el Síndrome de Carpenter, la enfermedad de Crouzon, el síndrome de Saethre-Chotzen y el síndrome de Pfeiffer que se caracterizan por presentar craneosinostosis, defecto congénito en el cual se produce un cierre anormal y precoz de las suturas craneales, provocando que la cabeza se deforme y con ello también la apariencia de la cara.
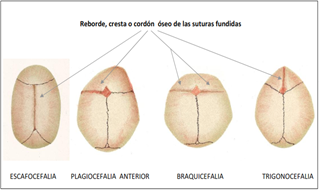
Descrito en 1906 por el médico francés Eugene Apert, presenta la misma distribución por sexos y número de casos: 0,11/10.000 nacimientos, y su génesis radica en mutaciones del gen principal FGFR2 que provocan una sobreexpresión de los genes EGFR y PDGRF-alfa. Se hereda de forma autosómica dominante, aunque la mayoría de los casos se deben a mutaciones de novo en parejas no afectadas, incrementándose el riesgo con la edad paterna.
Lo más característico por tanto de este síndrome es la fusión prematura del cráneo, lo que impide su crecimiento normal dando lugar a un hundimiento del tercio medio de la cara; no obstante, la variedad de síntomas anatómicos y viscerales es variable al igual que la presencia o no de retraso mental.
Podemos enumerarlas:
Alteraciones cráneo-faciales:
La escasa formación de la mitad de la cara se acompaña de ojos protruyentes, debido a una disminución del tamaño de la cavidad orbitaria. Los afectados pueden presentar lengua grande y maloclusión mandibular y en ocasiones paladar hendido y estrecho que les ocasiona infecciones frecuentes como otitis y afecciones respiratorias.
La deformidad de la bóveda craneal por cierre precoz de suturas que impide una expansión cerebral adecuada produce edema papilar, atrofia óptica, e incluso hipertensión intracraneal.
Manifestaciones músculo-esqueléticas:
Casi siempre sindactilias o fusión anormal de los dedos de las cuatro extremidades, así como fusión de vértebras cervicales, movilidad articular limitada, acortamiento de huesos largos como el fémur, húmero y el radio, hipoplasia de escápula, de pelvis, etc. Esto puede afectarles a la motricidad con andar torpe y necesidad de cirugía en la mayoría de los casos por las características de pies y manos.
Manifestaciones dermatológicas:
Pueden tener hiperhidrosis o exceso de sudoración, lesiones acneiformes máculo-pustulosas con áreas de hipopigmentación y engrosamiento de la piel.
Manifestaciones viscerales:
Aunque vinculadas a un retraso mental y psicomotor, cosa que no siempre ocurre en este síndrome, ocasionalmente pueden tener afecciones del sistema nervioso central que afectan al cuerpo calloso (agenesia o hipoplasia del mismo), con hipoplasia de la sustancia blanca.
Pueden aparecer alteraciones génito-urinarias como válvulas uretrales posteriores con resultado de hidronefrosis e insuficiencia renal en varones y en las niñas hipertrofia de clítoris.
En el aparato cardiocirculatorio suelen tener malformaciones cardíacas como la hipoplasia ventricular izquierda, comunicación interauricular y la coartación de aorta.
Características psicológicas
A causa de sus deformaciones físicas existe una descripción genérica de las características temperamentales y de conducta de las personas afectas de síndrome de Apert; hecho que depende de la existencia o no de retraso mental y del apoyo que reciban en el entorno familiar y social. En los niños sin retraso mental puede haber sentimientos de baja autoestima y miedo al rechazo; sentimientos que pueden estar atenuados en los niños con retraso mental asociado.
Suelen necesitar logopedas por sus dificultades en el lenguaje, pero expresan emociones con facilidad aunque su tendencia facial sea inexpresiva. Es frecuente detectar en estos niños dificultades de atención.
¿Puede diagnosticarse prenatalmente una craneosinostosis?
Dado que las personas afectas de este síndrome presentan cromosomas normales en el cariotipo y los niveles de a-feto proteína también se encuentran dentro de los límites normales, el diagnóstico antenatal solo puede confirmarse realizando un estudio genético de material fetal que demuestre la mutación en alguno de los genes específicos para la craneosinostosis, en el caso del Apert el gen FGFR2.
El diagnóstico prenatal puede hacerse en el primer o segundo trimestre de la gestación mediante técnicas moleculares de reacción en cadena de la polimerasa (PCR). La propia Asociación Nacional Síndrome de Apert lo describe en su página web.
1. Cuando existen antecedentes familiares de anomalías hereditarias, o bien un hijo anterior afecto de craneosinostosis, las muestras de células fetales para su procesamiento pueden obtenerse mediante biopsia corial entre las semanas 10 a 14 del embarazo.
2. En casos sin antecedentes el estudio se realiza cuando se observan signos ecográficos de sospecha, siendo los más frecuentes las deformaciones de la cara, de la bóveda craneal, la sindactilia de manos y pies y el aumento de líquido amniótico. En estos casos la obtención de células fetales se realiza de la semana 16 en adelante mediante amniocentesis y se procesan en laboratorio en un panel específico de perfil genético para craneosinostosis. O bien, si la sospecha no es concluyente para craneosinostosis y puede ser compatible con otras anomalías fetales de tipo malformativo, mediante estudio del Exoma[1].
[1] Estudio solo de una parte del genoma en donde se refleja el fenotipo o características anatómicas y morfológicas del individuo.